The Ehlers-Danlos Society – Why are people with EDS and HSD receiving such late access to innovative therapies?
What is Ehlers-Danlos syndrome and hypermobility spectrum disorders?
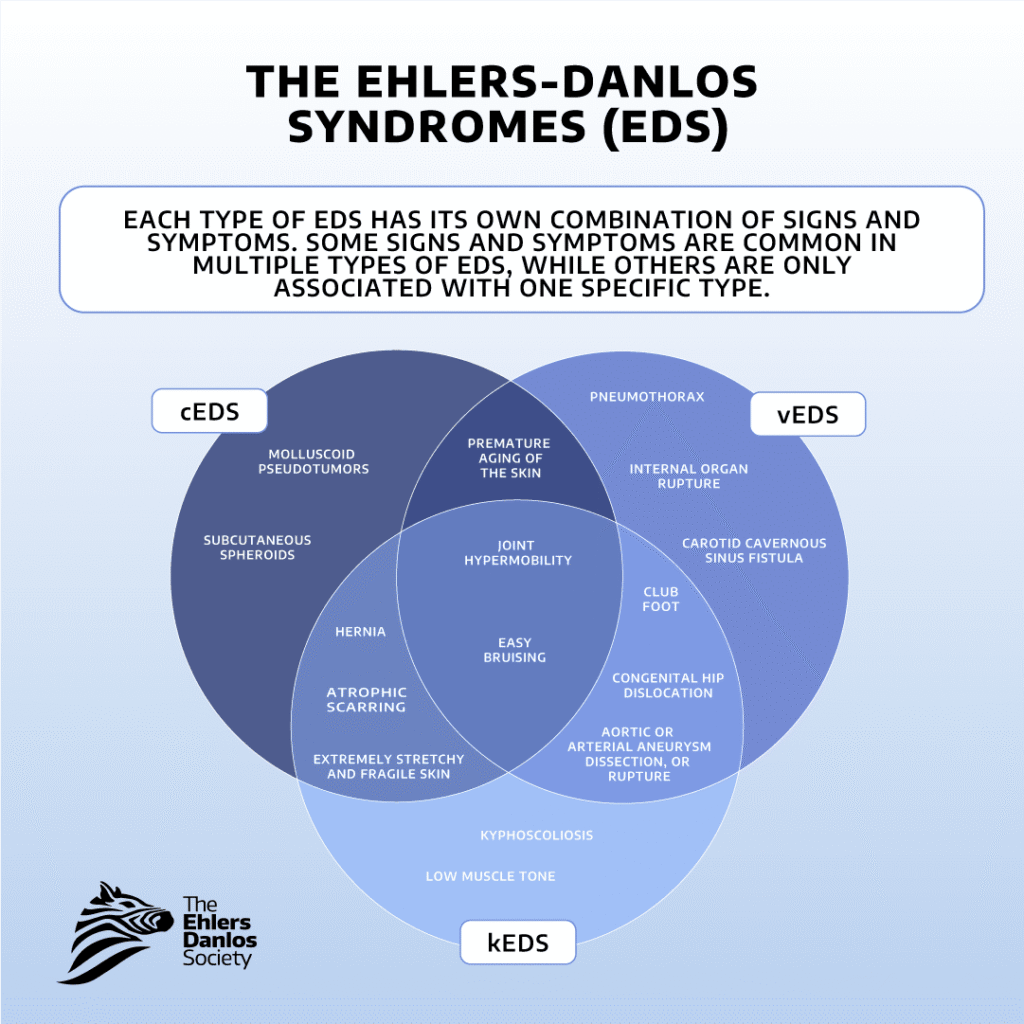
The Ehlers-Danlos syndromes are a group of heritable connective tissue disorders. Each type is caused by genetic variants that prevent connective tissue from functioning properly.
Twelve types of EDS have known genetic causes, and some types are associated with multiple different genes. The genetic cause(s) of hEDS have not been identified. Joint hypermobility, skin hyperextensibility, and tissue fragility are features seen across all types of EDS. HSD is diagnosed when someone has symptomatic joint hypermobility that can’t be explained by another condition. Many people with hypermobility spectrum disorders experience similar symptoms to those seen in hypermobile EDS. To receive a diagnosis of hEDS or HSD an individual will require ‘symptomatic hypermobility’ i.e. the joint hypermobility or instability they are experiencing leads to pain and/or injury. These symptoms may differ in severity, but for a diagnosis they cannot occur in isolation.

How EDS and HSD are treated
There is no cure for the Ehlers-Danlos syndromes, and treatment tends to focus on symptom management, via physiotherapy, pain management and orthopaedic devices. However, there are a number of medicinal therapies that can be used to help manage the wide range of symptoms associated with the disorders – many of which are overlooked. Vascular stress is a key component of some types of EDS and comorbidities like POTS, and drugs such as Beta-Blockers and celiprolol can help with managing blood pressure. There are a wide range of other drugs that can help with symptoms, from muscle relaxants to pain relief, gastrointestinal treatments, and even psychiatric medications to help with the mental health challenges that come from living with chronic pain and stress of such a complex disorder. But many patients are receiving treatments long after diagnosis, if they receive them at all. Here we look at the three key components identified by research that are causing that delay, along with the challenges that causes for patients – and what we can all do about them to improve the outlook.

1. The impact of diagnostic delay
Research consistently shows that people with Ehlers-Danlos syndromes (EDS) and hypermobility spectrum disorders (HSD) often experience long delays before receiving an accurate diagnosis. (Studies suggest that the time between first symptoms and diagnosis may exceed 10 years, and in some cases 15–22 years*1). These diagnostic delays can have a number of repercussions for patients’ lives. The severity of their pain commonly increases over time*2, and their quality of life typically decreases*3. Delays can also lead to higher rates of anxiety and depression*4 – all by-products that are being allowed to worsen. Another by-product is that patients lose faith in the healthcare systems supporting them*5, and even question the reality of the pain they are experiencing, due to repeated dismissal by under-informed professionals*6. The longer these symptoms go untreated the worse they become and the more ill and distrusting a patient is likely to be. If clinicians were able to recognise EDS and HSD sooner this would obviously help people access appropriate management strategies and supportive services sooner too, reducing their severity and dramatically improving their quality of life. But even when the condition and treatments are known, accessing drugs can still be a challenge. In some cases, access pathways do exist to help bridge this gap. Mechanisms such as early access programs, compassionate use, or named patient supply can enable patients to receive treatments before they are formally approved or widely available in their country. However, awareness of these pathways remains limited among both clinicians and patients, meaning opportunities for earlier access are often missed.
2. Challenges accessing treatments when EDS is not an approved indication
The fact that EDS and HSD don’t align neatly with certain drug types can mean that clinicians don’t know what to reach for when providing care. Specialist services or referral pathways are often limited, an issue exacerbated by fragmented healthcare systems and overburdened genetics services. Even when someone does obtain clinical support, limited knowledge of providers can mean those clinicians don’t know where to turn for innovative therapies that could help*7. In some healthcare systems, medications are only approved for specific indications. So if EDS and HSD are not on that list, patients can end up being unable to access therapies that could make the world of difference*8. Off-label management options for EDS and HSD are not generally discussed in medical literature, and this indicates that it isn’t so much that clinicians are facing barriers to prescription. It is more that there is a gulf in gaining access to that care. Critically it seems we need:
- Greater clinical awareness;
- Increased research into symptom management;
- And improved collaboration across healthcare, research, and industry
… in order to bridge that gap. It is the information and dialogue that is lacking. Improving visibility of available access pathways, and providing clearer guidance on how to navigate them, could play a key role in helping clinicians support patients more effectively – particularly in rare and complex conditions such as EDS and HSD.
3. Pain management limitations
Persistent pain is one of the most commonly reported symptoms among people with EDS and HSD. Because these conditions affect connective tissue throughout the body, pain can arise from a range of mechanisms, and vary widely between individuals. This means that there is no single management strategy that works for everyone. Management will usually involve finding bespoke combinations of several approaches, such as:
- Medication
- Movement-based therapies
- Physical rehabilitation
- Psychological approaches such as cognitive behavioural therapy
- Lifestyle adjustments
In some cases, small improvements from several strategies may collectively reduce pain and improve daily functioning. But the more personal the approach, the harder it is for clinicians to know which therapies to turn to in offering the right support.

The importance of continued research and collaboration
As with so many aspects of Medicines Access, information and awareness are absolutely critical in ensuring that patients are getting the right support. Continued research and collaboration between patient advocacy groups, clinicians, and industry are both essential in getting patients with EDS and HSD access to innovative therapies sooner. Because there are currently no treatments that address the underlying causes of EDS or HSD, management will continue to focus on addressing individual symptoms and associated risks with a targeted combination of approaches. As research continues our understanding of disease mechanisms for each type of EDS and HSD will improve, which will make it easier to identify potential therapeutic targets. Researchers should also be able to evaluate repurposed therapies for symptom management in people with EDS and HSD, but if we want to see progress all the different voices have to work together: researchers, healthcare professionals, patients, patient advocacy groups and industry partners. By working together we can accelerate innovation and improve care for people with EDS and HSD globally. This blog was written in partnership with the Ehlers-Danlos Society.
References
- *1 (Anderson & Lane, 2022; Halverson et al., 2023; Daylor et al., 2025; Wang et al., 2024)
- *2 (Besson et al., 2020; Kalisch et al., 2019)
- *3 (Anderson, 2026)
- *4 (Halverson et al., 2023)
- *5 (Terry et al., 2015; Bulbena et al., 2017)
- *6 (Halverson et al., 2025)
- *7 (Anderson & Lane, 2022; Carbone et al., 2025; Dar et al., 2020; Dockrell et al.,
- 2021; Knight et al., 2022; Wagner et al., 2024)
- *8 (Dar, 2021)